Performance evaluation is a critical requirement for in vitro diagnostic (IVD) devices and forms part of the technical documentation necessary for placing an IVD device on the EU market. The requirements for performance evaluation are described within Chapter VI of the Regulation on in vitro diagnostic medical devices (EU 2017/746) (IVDR) and supported by Annexes I, II, III and XIII.
Understanding Performance Evaluation: What You Need to Know
Chapter VI of the IVDR states:
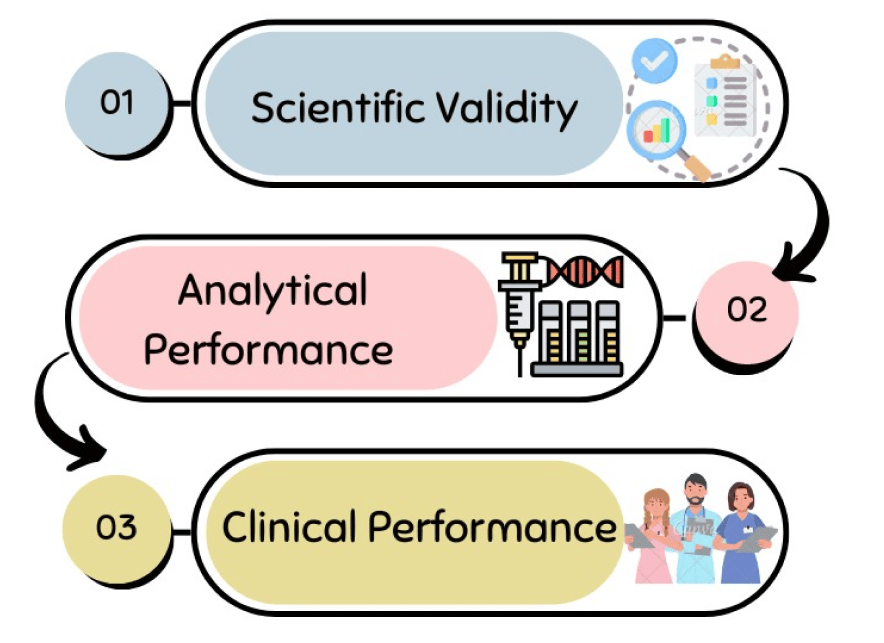
“A performance evaluation shall follow a defined and methodologically sound procedure for the demonstration of the following, in accordance with this Article and with Part A of Annex XIII:
- scientific validity;
- analytical performance;
- clinical performance;
The clinical evidence derived from the performance evaluation shall provide scientifically valid assurance that the relevant general safety and performance requirements (GSPRs) set out in Annex I are fulfilled under normal conditions of use.
A Closer Look at Article 56
Article 56 of the IVDR refers to the GSPRs set out in Annex I, specifically relating to the performance characteristics described in Chapter I and Section 9 of Annex I. It also addresses the acceptability of the benefit-risk ratio, as outlined in Sections 1 and 8 of Annex I, which is based on the scientific validity, analytical performance, and clinical performance of the IVD. Annex XIII specifies how IVD manufacturers must plan, conduct, and document the performance evaluation.
Manufacturers are required to establish a standard operating procedure (SOP) for IVD performance evaluations, ensuring that it covers the entire device lifecycle. The IVDR defines performance evaluation as a continuous process that closely interfaces with risk management, as referenced in Annex VII, Section 4.5.4, and Annex XIII, Section 1.1.
Mastering the Performance Evaluation Plan
A device-specific Performance Evaluation Plan (PEP) must be prepared in accordance with Annex XIII, Section 1.1 of the IVDR. The PEP should outline the current state of the art in relation to the medical, diagnostic, and technology-related fields relevant to the device. It must also reference applicable standards, common specifications, guidelines, or best practice documents.
The PEP should define the intended purpose and characteristics of the device, including the analyte or biomarker it detects, its intended use, target user, and indications for use. Additionally, it must outline how the device demonstrates compliance with the GSPRs through its performance evaluation. The plan should also briefly describe the methods used to assess the device’s analytical and clinical performance, benefit-risk considerations, and the Post-Market Performance Follow-up (PMPF) plan.
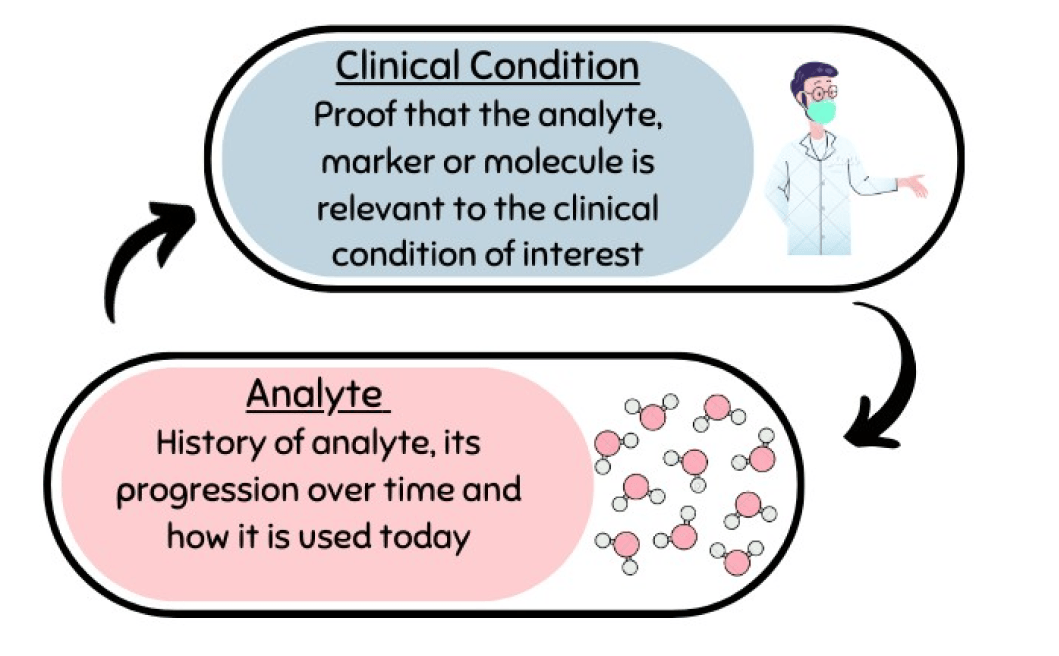
Scientific Validity Explained
Scientific validity refers to the association between the analyte measured by the device and the clinical condition or physiological state. This association is documented in a Scientific Validity Report (SVR). The SVR should establish the device as state-of-the-art and demonstrate its clinical benefit. It should provide evidence of the clinical background, the clinical condition, the significance of the analyte, currently-used diagnostic options, similar and/or equivalent devices, and recommendations from professional societies to support the IVD’s performance and intended use.
Navigating Analytical Performance
Section 9.1 of Annex I outlines the analytical performance requirements. However, as it is not possible to anticipate all scenarios regarding device types and intended purposes, the responsibility lies with the manufacturer to provide a rationale for any characteristics that are not applicable to their device. EN ISO 18113-3:2022, In vitro diagnostic medical devices — Information supplied by the manufacturer (labelling) — Part 3: In vitro diagnostic instruments for professional use, offers guidance on performance characteristics that should be included in the labelling for instruments. Analytical performance must always be demonstrated through analytical performance studies, either using existing data or generating new evidence.
Clinical Performance Uncovered
Clinical performance is the “ability of a device to yield results which are correlated with a defined clinical/physiological/pathological condition or state following the target population and intended user”. Clinical performance provides evidence of how effectively the IVD device delivers results, diagnoses, or guides treatment decisions. Annex XIII, Section 1.2.3 of the IVDR outlines multiple options for gathering clinical performance data to substantiate the clinical evidence of the device.
For devices measuring analytes that are associated with a clinical condition that have medical decision points, clinical performance data is required and should form part of the clinical performance report (CPR). Typical clinical performance data could be diagnostic sensitivity and specificity, area under the curve, negative predictive value, and positive predictive value.
For devices measuring analytes without clear medical decision points or for devices measuring analytes that are not (yet) associated with a clinical condition, clinical performance may be defined as correlation with a physiological state, or a justification for omission of clinical performance studies may be considered. Typical data presented in the CPR would be negative percent agreement and positive percent agreement.
Demonstration of the clinical performance of a device is based on one or a combination of the following sources:

The Key Steps in Crafting a Performance Evaluation Report
In the Performance Evaluation Report (PER), you will need to summarise all the results related to Scientific Validity, Analytical Performance, and Clinical Performance to demonstrate conformity with the relevant GSPRs for your device, as referred to in Annex I of the IVDR. In the PER, you should evaluate the clinical evidence in light of the current state of the art in medicine, technology, the intended purpose, and the device’s positive benefit-risk ratio.
Annex VIII, Part A (1.3.2) of the IVDR outlines the specific components of the PER and specifies that it must include:
- the justification for the approach taken to gather the clinical evidence;
- the literature search methodology and the literature search protocol and literature search report of a literature review;
- the technology on which the device is based, the intended purpose of the device, and any claims made about the device’s performance or safety;
- the nature and extent of the scientific validity and the analytical and clinical performance data that has been evaluated;
- the clinical evidence as the acceptable performances against the state of the art in medicine;
- any new conclusions derived from PMPF reports
PERs for Class C and D devices must be updated at least annually, whereas PERs for Class A and B devices should be updated as needed, although at least a three-year review cycle would be recommended. As this document is multi-disciplinary, it is crucial that the manufacturer organises the writing and maintenance of this document and fully integrates it into the infrastructure of the quality management system (QMS).
Ensuring Ongoing Success with Post-Market Performance Follow-Up
Post-Market Performance Follow-Up (PMPF) is defined in Part B of Annex XIII as ‘a continuous process that updates the performance evaluation’ and goes on to state ‘with the aim of confirming the safety, performance and scientific validity throughout the expected lifetime of the device, of ensuring the continued acceptability of the benefit-risk ratio and of detecting emerging risks on the basis of factual evidence’.
PMPF essentially serves to continue supporting the PEP/PER throughout the device’s lifecycle and is particularly useful for monitoring scientific or clinical developments that may impact the device’s performance and safety.
Outputs from PMPF are written in the PMPF evaluation report, where it is then determined if those outputs result in:
- An update to the associated performance evaluation documentation and a revision of the risk management reports
- A CAPA being raised
- Further PMPF studies being conducted
Ensuring IVD Success: Key Takeaways for Performance Evaluation
Conducting a performance evaluation for IVDs is a critical step in ensuring their safety, reliability, and effectiveness. By following a structured process—starting with understanding regulatory requirements, defining intended use, and conducting rigorous analytical and clinical testing—you can demonstrate that your IVD meets the necessary standards for market approval. Continuous post-market surveillance further ensures that the device continues to deliver value and contributes to better patient outcomes.
At Mantra Systems, we simplify the process, ensuring your device meets all regulatory requirements for EU market access. Let us take the complexity out of performance evaluation so you can focus on delivering safe, effective solutions. Book a call today.