Post-Market Clinical Follow-up (PMCF) of medical devices
A guide to PMCF under the Medical Device Regulation (MDR) 2017/745
What is Post-Market Clinical Follow-up (PMCF)?

Post-Market Clinical Follow-up is an important component of Post-Market Surveillance (PMS) that applies to almost all medical devices under the EU MDR.
It is a proactive process that collects and evaluates clinical data on the safety and performance of a medical device in normal use. According the MDR Annex XIV Part B, PMCF should run on a continuous basis throughout the entire lifetime of a device.
PMCF has been an important component of medical device regulatory frameworks since the introduction of the Medical Device Directive (MDD). However, the introduction of the MDR elevated the importance of PMCF and, with very limited exceptions, it is now required for all medical devices regardless of risk class.
It is vital that medical device manufacturers have a solid understanding of PMCF requirements under the EU MDR regulatory framework.
What are the objectives of PMCF?
According to Annex XIV Part B of the MDR, overall objectives of PMCF are to collect and evaluate clinical evidence to assess the safety and performance of a medical device when used as intended. Specific objectives for PMCF under the MDR include:
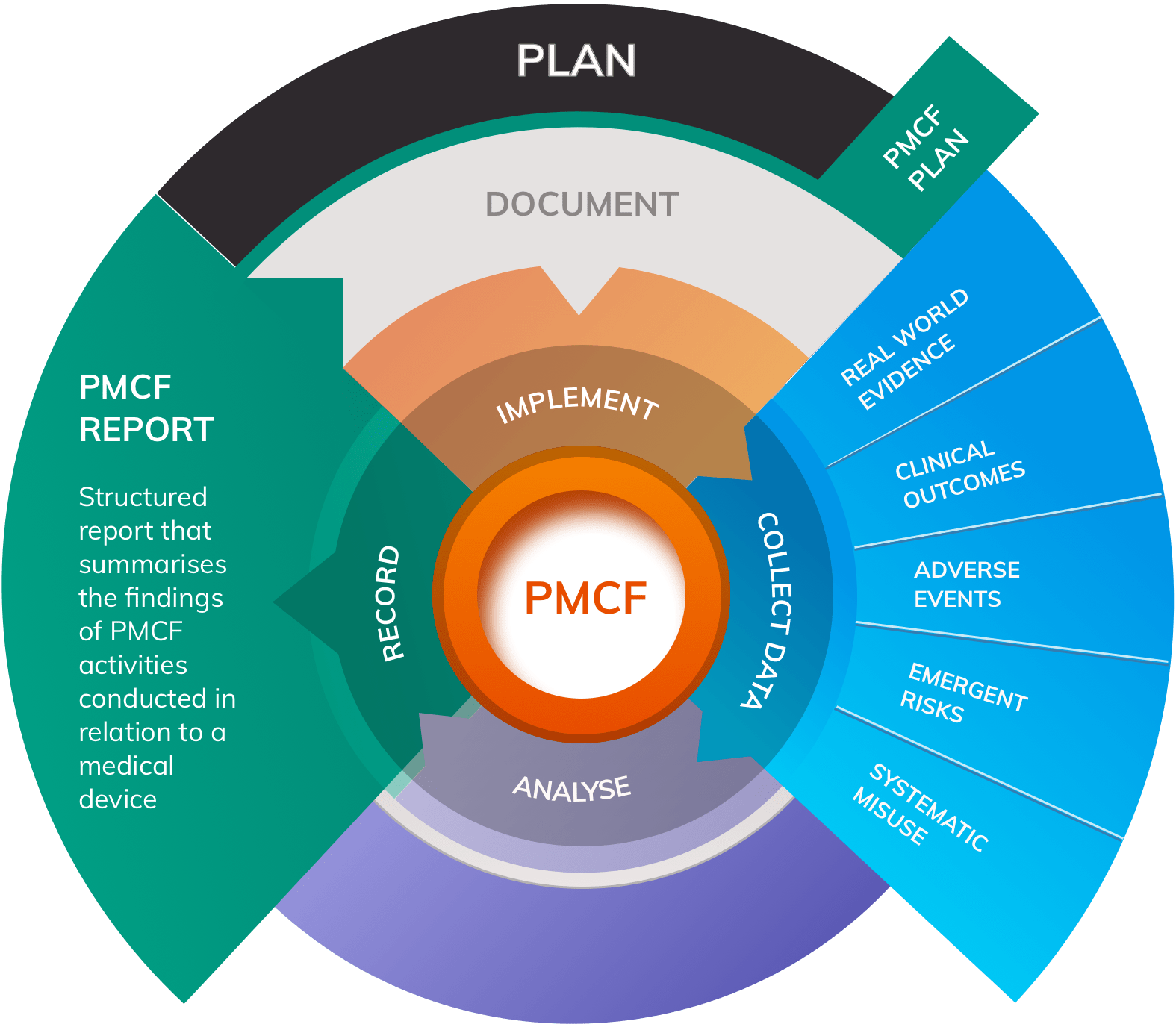
- Monitoring frequency and severity of residual risks known to be associated with the device
- Contributing towards the update of Clinical Evaluation
- Detecting any new or emergent risks and previously unknown side-effects
- Confirming the overall safety and performance of the medical device in normal use
- Identifying systematic misuse of the device and its impact on safety and performance
The design of PMCF studies and surveys should be documented in a PMCF Plan and results must be collated in a PMCF Report that forms an important input to the Clinical Evaluation Report (CER) for the device.
A PMCF study or survey must be designed to accommodate the characteristics, target patient and user population, risk classification and complexity of the subject device, meaning that experience in designing clinical investigations is vital in ensuring suitability for purpose.
Design considerations for PMCF under the MDR
Manufacturers should adopt a strategic approach when designing a PMCF system to meet MDR requirements, often meaning that dedicated types of clinical investigation are used to meet the full range of requirements. Most ‘standard’ clinical investigations collect data only for a limited time and on a limited patient population, meaning that they may fail to meet PMCF requirements for longitudinal data collection throughout the entire lifetime of a device.

Designing PMCF to produce Real World Evidence (RWE) on device safety and performance in normal use is a powerful approach to PMCF under the MDR. Real World Evidence:
- Captures data from a study / survey population that represents the entire population normally exposed to the device
- Is non-comparative, focusing only on the safety and performance of the subject device rather than making comparisons between different devices
- Does not involve experimental exposure; rather, it studies the use of a device that is already CE-marked and seeks to confirm or refute that it meets the necessary safety and performance requirements.
- May be derived from a registry that, in principle, recruits an uncapped number of patients and runs indefinitely throughout the device’s lifetime
- Is conducted at all types of clinical site and does not select for centres of excellence of other sites that may be unrepresentative or positively biased towards better results
Several categories of PMCF systems are capable of generating RWE of the required standard, with a PMCF study being the most versatile and powerful design. For lower-risk or consumer devices, a PMCF survey can be an effective and low-cost strategy.
Well-designed Clinical Evidence Generation systems are heavily influenced by the findings of Risk Management, incorporating a design that ensures the ongoing collection of data to demonstrate benefit-risk acceptability throughout a device’s presence on the market.
PMCF Clinical Investigations - documentation and considerations
Clinical Investigations (including medical device registries and PMCF surveys) must adhere to all legislative requirements related to the performance of medical research.
Specific requirements for designing, conducting and reporting Clinical Investigations are documented in MDR Annex XV. Investigations must also conform with GDPR requirements on data collection and should meet the standards of Clinical Investigation conduct outlined in ISO 14155:2020 and GCP guidelines.
For all PMCF investigations, documents should include:
- A Clinical Investigation Plan (CIP) or protocol that outlines how the PMCF study must be conducted
- An Investigator’s Brochure that specifies responsibilities for the principal investigator at each clinical site
- A patient information leaflet and consent form that ensures patients are fully informed and documents the process of informed consent
Furthermore, consideration must be given to how data will be collected and stored. Many manufacturers opt for eCRF and ePRO systems that facilitate secure data collection and storage from any internet-enabled device.
What are the requirements for developing a PMCF Plan?
Creating a suitable PMCF Plan requires a combination of regulatory experience, clinical expertise, and familiarity with relevant legislation and guidelines. Although Annex XIV Part B of the MDR outlines the substantive requirements of PMCF systems, being aware of and working with appropriate supporting guidelines can be of great assistance in producing an MDR-compliant PMCF strategy.
One such guideline is MDCG 2020-7 that provides a template for producing MDR-compliant PMCF Plans. It also contains guidance on structuring PMCF objectives and helps ensure that PMCF design incorporates the full range of elements required for MDR compliance.
Additionally, the MedDev range of guidelines can be invaluable in meeting the requirements of the MDR. The relevant guideline for PMCF is MedDev 2.12/2 rev 2. This MedDev guideline covers a range of topics including:
- When a PMCF study is indicated
- What is required in a PMCF study
- How to use study data
- The role of Notified Bodies in working with PMCF data
MedDev 2.12/2 rev 2 was produced in reference to requirements under the out-going Medical Device Directive MDD 93/42/EC and has not yet been updated to meet the specific requirements for PMCF imposed by the MDR. Nonetheless, it serves as a very useful starting point in designing a PMCF strategy.
Refining the guidance in MedDev 2.12/2 rev 2 into an MDR compliant PMCF strategy requires clinical expertise, an understanding of how to drive subject recruitment at clinical sites, and detailed familiarity with the MDR requirements for designing clinical investigations in Annex XV.
It also necessitates a high level of understanding of handling subject data to meet the obligations imposed by GDPR, and construction of a secure data handling and storage system.
How does PMCF relate to Post-Market Surveillance (PMS)?

PMCF is a component of Post-Market Surveillance (PMS) and it is important to understand that the two systems, while related and inter-dependent, are not the same.
An effective medical device PMS system will consist of a PMCF system that proactively and continually collects data on normal use, and a Vigilance system that gathers information relating to complaints and adverse events, and that handles FSCAs and FSNs.
While PMCF will always return some data, it is theoretically possible that a Vigilance system would return no data — for example, if the device worked flawlessly and resulted in no complaints, adverse events or serious incidents.
PMS, in turn, is a device-specific system that is part of the organisation’s Quality Management System (QMS) that oversees quality across full range of company activities.


