PMCF Plans
How to write a PMCF Plan for medical devices under the Medical Device Regulation (MDR) 2017/745
What is a PMCF Plan?
A PMCF Plan is an important technical document that outlines a method for conducting Post-Market Clinical Follow-up on a medical device.
A PMCF Plan must be submitted for regulatory approval of devices under the EU MDR as a component of the MDR Annex II technical documentation.
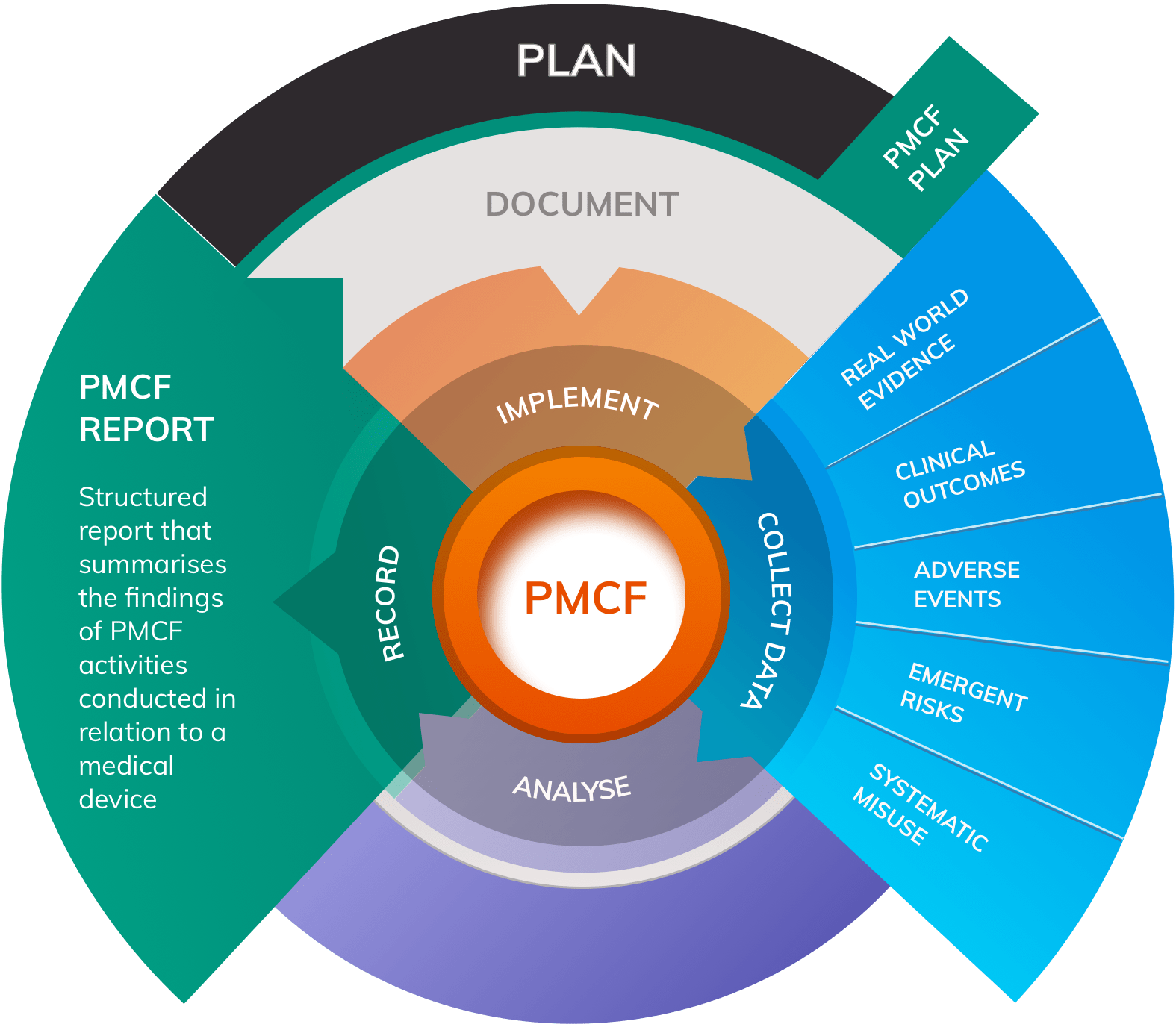
MDR Annex XIV Part B requires that a PMCF Plan must provide a detailed summary of a method that will:
- Confirm safety and performance of the device throughout its expected lifetime
- Identify previously unknown side-effects
- Monitor the rate and severity of known side-effects
- Identify and analyse any emergent risks
- Ensure continued benefit-risk acceptability
- Identify any systematic misuse or off-label use
Writing a PMCF Plan is a process that draws upon expertise in technical writing and clinical investigation design.
How to write a PMCF Plan
Writing a PMCF Plan involves two stages:
- PMCF system design - PMCF system must be designed to meet the needs of the subject device before a PMCF Plan can be written.
- PMCF Plan structure - once designed, the PMCF system must be documented in a PMCF Plan that is structured according to MDR requirements.
1. PMCF system design
PMCF system design should be tailored to the subject device and its associated classification and complexity. Effective PMCF systems are built to address risk management outcomes alongside the overall objectives for PMCF as specified in the MDR. A PMCF system must be capable of capturing data on use of a medical device throughout its entire lifetime, meaning that many standard-type clinical investigations will not be suitable for this application.
PMCF systems can range from simple patient or end-user surveys to formal clinical investigations and complex medical device registries that incorporate both clinical and patient-feedback data. All clinical investigations must adhere to MDR Annex XV requirements on design, documentation, consent and data-handling, as well as external legislation such as the GDPR and standards such as ISO 14155:2020.
Increasingly, manufacturers are advised to develop a PMCF Plan that outlines a method for generating Real World Evidence (RWE) that provides data on real, undifferentiated use of the product in the hands of genuine users “out there in the world”. Standard clinical investigations tend to be too restrictive to meet requirements for PMCF under the MDR and will not run for a sufficient length of time to meet requirements for longitudinal data collection.
A medical device registry is a data collection system that provides a single central repository of PMCF data captured from any number of different territories. Properly designed, a PMCF study/survey/registry ensures that data is generated in a consistent manner under a universal study protocol. It also ensures adherence to wider rules and regulations including the requirements for clinical investigations in Annex XV MDR.
Clinical Evidence Generation systems require specialist knowledge to design and implement. Once established, a well-designed system can pay huge dividends through ensuring ongoing availability of data to support regulatory approval of a medical device throughout its commercial lifecycle.
2. PMCF Plan structure
The PMCF Plan must comprehensively outline the systems and processes through which the PMCF system will function. The PMCF Plan will be subject to regulatory review from Notified Bodies and other regulatory authorities alongside other technical documentation.
A well-written PMCF Plan should include at least:
- Details of general methods to be applied, e.g. gathering clinical data, user feedback and/or literature searching
- Specific methods to be used, e.g. PMCF study/survey/registry
- Rationale for selected general and specific methods
- A reference to relevant parts of the Clinical Evaluation Report (CER) that contain a summary of PMCF data, an appraisal of the PMCF system, and results of data analysis.
- Specific objectives addressed by PMCF
- Evaluation of clinical data from equivalent/similar devices, if relevant
- Reference to any harmonised standards used in developing the PMCF Plan
- Timescale of PMCF activities
While not obligatory, it is advisable to adopt the PMCF Plan structure recommended in MDCG Guideline 2020-7 which ensures that all MDR Annex XIV Part B requirements have been met.
Post-Market Clinical Follow-up (PMCF) under the MDR
PMCF is the process of proactively and constantly collecting clinical data in order to demonstrate that a CE-marked medical device’s safety and performance is in line with expectations.
The Medical Device Regulation (MDR) 2017/745 greatly enhances the importance of PMCF compared to the MDD. A detailed set of requirements for the design and conduct of PMCF activities is outlined in MDR Annex XIV Part B. A specific requirement is that all manufacturers document a PMCF Plan to detail the process that will be followed in collecting PMCF data on each of their medical devices.
It is important to account for all relevant laws and regulations when developing a PMCF Plan. In particular, the General Data And Performance Regulations (GDPR) establishes a strict set of standards for handling study subject’s clinical data, and the PMCF Plan should establish procedures for ensuring that the requirements of this Regulation are adhered to.
Furthermore, it is important to consider whether PMCF activities may require ethical approval before data collection can commence. Requirements for ethical approval vary between different EU Member States so it is important to understand local requirements and ensure that the PMCF Plan incorporates procedures for meeting obligations. Mantra Systems can help you understand this process, so speak with a member of our team today.


