Creating a usability file is a crucial part of the product development process for medical devices, including Software as a Medical Device (SaMD), to ensure user-centred design and safety. IEC 62366-1 provides a structured approach to ensure that devices are designed with the user in mind and that potential risks associated with their use are identified and mitigated.
What is IEC 62366? A Crucial Standard for Medical Device Usability
According to ISO, ‘IEC 62366 specifies a process for a manufacturer to analyse, specify, develop and evaluate the useability of a medical device as it relates to safety. The first edition of IEC 62366-1, together with the first edition of IEC 62366-2, cancels and replaces the first edition of IEC 62366 published in 2007 and its Amendment 1 (2014)
’
- IEC 62366-1 — Application of usability engineering to medical devices
- IEC 62366-2 — Guidance on the application of usability to engineering to medical devices
This guideline provides medical device manufacturers with a structured approach to integrate human factors, supporting them in assessing, defining, creating, and testing the usability of their medical devices.
Navigating the Process: Key Steps in Writing Your Usability File
What exactly should be included in a Usability Engineering File? How can you ensure that all the requirements outlined in the standard are met? What steps do you need to take to ensure compliance? These are common questions that many people face when beginning to create their usability files. By understanding the essential components and requirements, you’ll be well-equipped to develop a comprehensive Usability Engineering File that meets all necessary standards and helps ensure your medical device is both safe and effective for use.
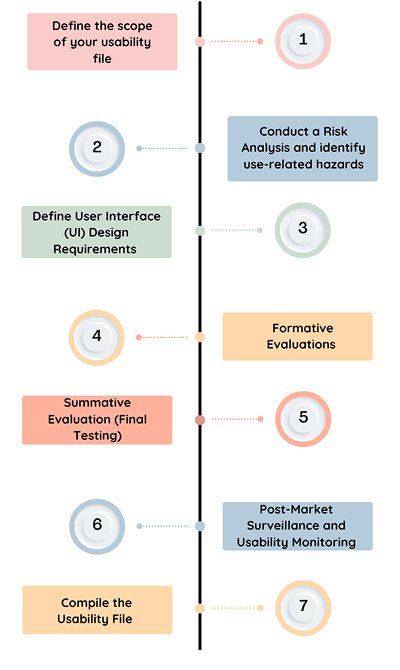
Step 1: Define the Scope
This encompasses the specific user group, the intended purpose, and the context in which the device will be utilised.
Intended Users: Who will be using the device? This could include healthcare professionals, patients, or other relevant parties.
Intended Use: What is the device designed to accomplish? This refers to the specific tasks the device is intended for, such as monitoring, diagnosing, or therapeutic applications.
Use Environment: Where will the device be used? The setting can significantly influence the device’s usability, whether it’s intended for use in a home environment or a clinical/hospital setting.
Step 2: Conducting a Risk Analysis
IEC 62366-1 mandates that you perform a comprehensive risk analysis to identify potential use-related hazards—situations where user errors may lead to harm or malfunction of the device.
Hazard Identification: Identify potential hazards that may result from user errors or design flaws. For example, in a medical device like a ventilator, if the user interface is poorly designed, a healthcare professional could mistakenly adjust the settings, leading to inappropriate ventilation rates for the patient. This could result in either hypoventilation (insufficient oxygen delivery) or hyperventilation (excessive oxygen delivery), both of which could cause severe harm to the patient.
Risk Assessment: Assess the likelihood and severity of harm associated with each identified hazard. This involves evaluating whether a specific user action could result in patient harm, an incorrect diagnosis, or delayed treatment, ensuring that risks are appropriately managed and mitigated.
Step 3: Define User Interface (UI) Design Requirements
Following the analysis of user needs and risks, the next crucial step is to establish UI design requirements tailored to the target user group. Chapter 5.2. requires you to identify, as part of the risk analysis, all parts and characteristics of the User Interface that are related to the safety of the medical device.
User Capabilities: Consider the cognitive, physical, and sensory abilities of the users. For instance, if the device is intended for elderly patients, ensure the interface is intuitive, with large text and simplified navigation.
Design Guidelines: These should focus on clear labelling, well-organised controls, alarms, and instructions for use (IFU). Specific adaptations may be necessary for certain groups, such as non-professional users or individuals with visual impairments.
Task Optimisation: Structure the UI to reduce the likelihood of user error. Important tasks should be straightforward to access and perform, eliminating unnecessary steps that could lead to mistakes.
Step 4: Formative Evaluations
The formative evaluation section involves the testing of early prototypes or even design concepts with users in order to identify any potential problems prior to the final design being confirmed and completed. Once risks are identified, this process will repeat until these are mitigated.
User Testing: Carry out usability testing with representative users to assess the device’s interface and interaction design. This could involve techniques such as cognitive walkthroughs, task analysis, and heuristic evaluations to identify potential issues and areas for improvement.
Were critical use errors found? Are further product improvements necessary? If you have answered “yes” to either of these questions at this point, go back as far as necessary in the Usability Engineering process and iterate through it from this point on until no new and critical problems occur during use. If this is the case, you are ready for the last big step: performing the summative evaluation.
Step 5: Summative Evaluation
The goal is to prove that the medical device (and its Interface) can be used without unacceptable residual risks for users, patients, and third parties. The hazard-related use scenarios previously defined are tested.
The summative evaluation focuses on validating the final design to ensure that the device meets the defined usability goals and operates safely and effectively in real-world conditions.
Final Usability Testing: Conduct usability testing on the final version of the device to confirm that it functions as expected and that users can interact with it safely. This typically involves more realistic use scenarios, which may take place in clinical or home environments.
Task Performance Metrics: Evaluate whether users can complete key tasks successfully, such as using the device without errors, interpreting information accurately, and following safety instructions correctly. These metrics help ensure the device’s usability and overall safety in practical use.
Step 6: Post-Market Surveillance (PMS) and Usability Monitoring
Once the product has been released to the market, ongoing monitoring of its usability is crucial. Post-market surveillance helps to identify any emerging issues or areas where improvements can be made.
Incident Reporting: Set up a system to collect feedback and reports related to user issues. This should include complaints, adverse events, or any usability failures encountered by users.
Usability Improvements: If usability issues are identified after the product has been launched, they should be addressed through iterative improvements or design updates to enhance performance and user experience.
Step 7: Compiling the Usability File
The Usability Engineering File should provide a thorough and organised record of the entire usability process, from initial risk analysis and design specifications to testing and post-market surveillance. It is essential that the file demonstrates full compliance with the usability requirements of IEC 62366-1, showing that every step has been taken to guarantee the device’s safety and effectiveness.
The file should include:
- The device’s scope and intended use
- A detailed hazard analysis and risk assessment
- Clear design specifications and user interface requirements
- Results from formative and summative testing
- A post-market plan for ongoing usability monitoring
Your Path to IEC 62366 Compliance: Key Takeaways
The Usability Engineering process, as meticulously outlined in IEC 62366-1, plays a crucial role in ensuring the safety and effectiveness of medical devices. By closely integrating with Risk Management, this process helps manufacturers identify and address potential use-related hazards that could jeopardise user safety, patient health, or device performance. Effective implementation, alongside comprehensive documentation, is not only vital for regulatory compliance but also for minimising the risks associated with human error. Ultimately, the goal is to create devices that are intuitive, safe, and reliable and that can stand up to real-world challenges. Following the steps of the Usability Engineering process ensures that these objectives are met and that safety is prioritised in every phase of development.
Standards like IEC 62366-1:2015 are just one part of the increasingly complex regulatory landscape facing medical device companies. At Mantra Systems, we specialise in helping you navigate these challenges with clarity and confidence.
Whether you’re preparing a technical file, responding to questions from a notified body, or working toward approval, our team provides practical, tailored support at every stage. We offer strategic guidance across regulatory frameworks, assist with technical documentation, and help you stay aligned with evolving requirements.
If you’re looking for a trusted partner to take the pressure out of compliance, get in touch with Mantra Systems today.